
Overconfident: What Structure Prediction Confidence Scores Tell Us About Binding
by Joseph Harman and Natasha Murakowska

At a Glance: Computational protein design pipelines used in early drug development rely on structure prediction confidence scores to select which designs get tested in the lab. But how well do these scores generalize across diverse protein engineering tasks, particularly for antibody–antigen systems? Because these metrics shape real drug development decisions about which molecules to test and which to discard, it is critical to understand when they are reliable and when they are not.
Here, we examine the relationship between Boltz-2 confidence scores and experimentally measured binding affinity for AlphaSeq datasets spanning tens of thousands of VHHs, scFvs, native and engineered antigens, de novo minibinders, and mutational scans totaling >7 million quantitative binding affinity measurements. We evaluate the impact of templating during prediction and analyze how confidence scores perform across distinct design scenarios. Our findings reveal that confidence scores work best as negative filters: they effectively remove many non-binders. However, they fail to reliably rank affinity, produce high false positive rates, and especially struggle to capture the effects of point mutations. Moving beyond filtering will require explicitly training models on large, quantitative datasets that span both broad sequence diversity and fine-grained mutational landscapes.
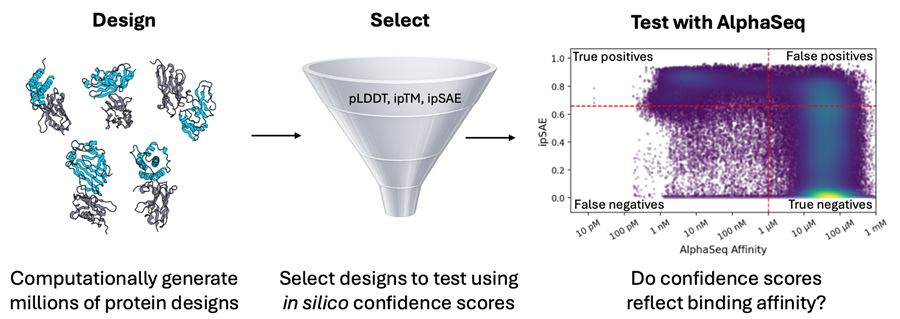
Confidence Scores are Central to Modern Protein Design Workflows. But How Well do they Work?
Structure prediction models such as AlphaFold, RosettaFold, and Boltz-2 are key elements of de novo protein design workflows, both for generating candidates and ranking them for experimental testing. A common strategy for these design approaches is to optimize and filter on interface-focused confidence scores with the goal of enriching true binders [1–4].
Current de novo design methods [5–10] extend this approach through integrated docking and sequence proposal strategies, upgraded structure prediction models, and expanded filtering criteria. Many methods explicitly optimize interface confidence scores, such as interface predicted TMscore (ipTM) and interface predicted score from aligned errors (ipSAE) [11]. Retrospective analyses of minibinder [12] and antibody [9] datasets consistently rank these metrics among the strongest computational predictors of binding, and ipSAE remains the primary screening metric for wet-lab validation in recent protein design competitions [13].
Despite their widespread use, the predictive value of confidence scores is more limited than their prevalence suggests. Recent work has begun to quantify this: in the Adaptyv EGFR competition, ipTM achieved an AUROC of 0.64 for binding classification, leading the authors to conclude that these metrics were “insufficiently predictive of true experimental binding probability or affinity” [13]. We observed a high false positive rate for ipSAE in an AlphaSeq dataset of inverse-folded VHH variants. Point mutation campaigns are a particular blind spot: AlphaFold is known to poorly capture the energetic effects of point mutations [14], and metrics like ipSAE have not been systematically evaluated on point mutation data. Quantitative datasets at the scale of AlphaSeq—spanning tens of thousands of measurements across diverse binder formats, engineered antigens, and mutation-rich campaigns—offer an opportunity to characterize more precisely where confidence metrics succeed and where they fail.
We ask two main questions in this analysis:
How robust are interface confidence scores across native, engineered, and de novo PPIs, particularly for antibody-antigen systems?
How do structure prediction variations, such as providing a template structure during inference, impact the performance of confidence scores?
To answer these questions, we generated Boltz-2 complex predictions for tens of thousands of known and designed PPIs with experimentally measured AlphaSeq affinities. These datasets included de novo minibinders and antibodies, wildtype and engineered antigens, and mutation-rich optimization campaigns, each representing a distinct protein design use case (Table 1). Our previous blog posts describe our inverse folding and SabDab-nano (SDN) point mutant datasets. The SEPIA datasets are part of a novel “synthetic antigen” data generation approach that we will discuss in greater detail in a forthcoming preprint; briefly, SEPIA datasets consist of de novo minibinders designed to bind to VHH paratopes.
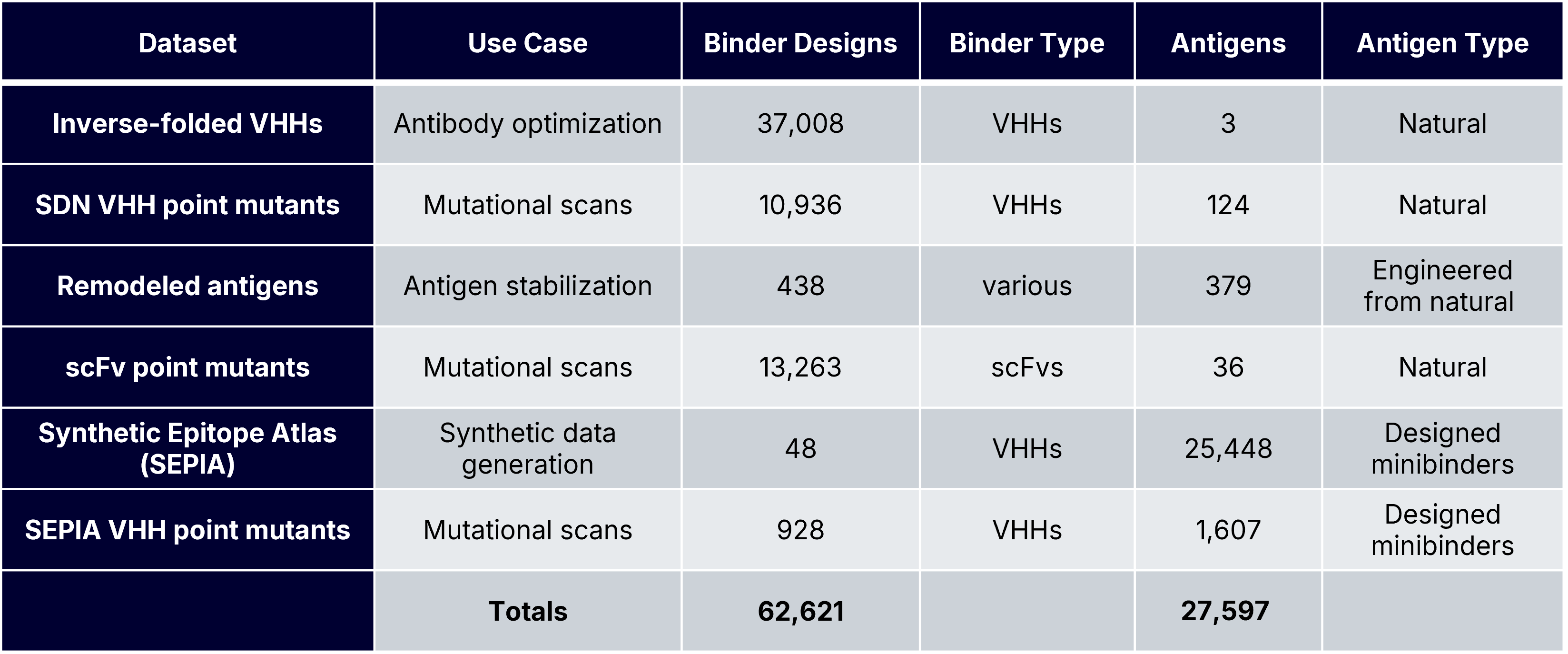
Structure Prediction and Evaluation Framework
To produce confidence scores for each measured PPI, we used Boltz-2 to predict three complex structures (diffusion samples) without generating multiple sequence alignments. We also investigated the effect of providing different structural inputs (templates) during Boltz-2 complex prediction, as previous work shows that providing different structural priors during complex prediction affects agreement between confidence scores and experimental success [1,3]. We examined three different templating strategies:
No templates – Provide only the sequences of the binder and antigen.
Soft templating – Provide the full complex structure.
Hard templating – Provide the complex and constrain predictions within a distance threshold (3Å) of the supplied template.
Binder-antigen co-structures were supplied as templates for all datasets except for the scFv point mutant dataset, which lacked a solved antibody-antigen co-structure. For the scFv dataset, the antigen monomer structure was provided for both soft and hard templating.
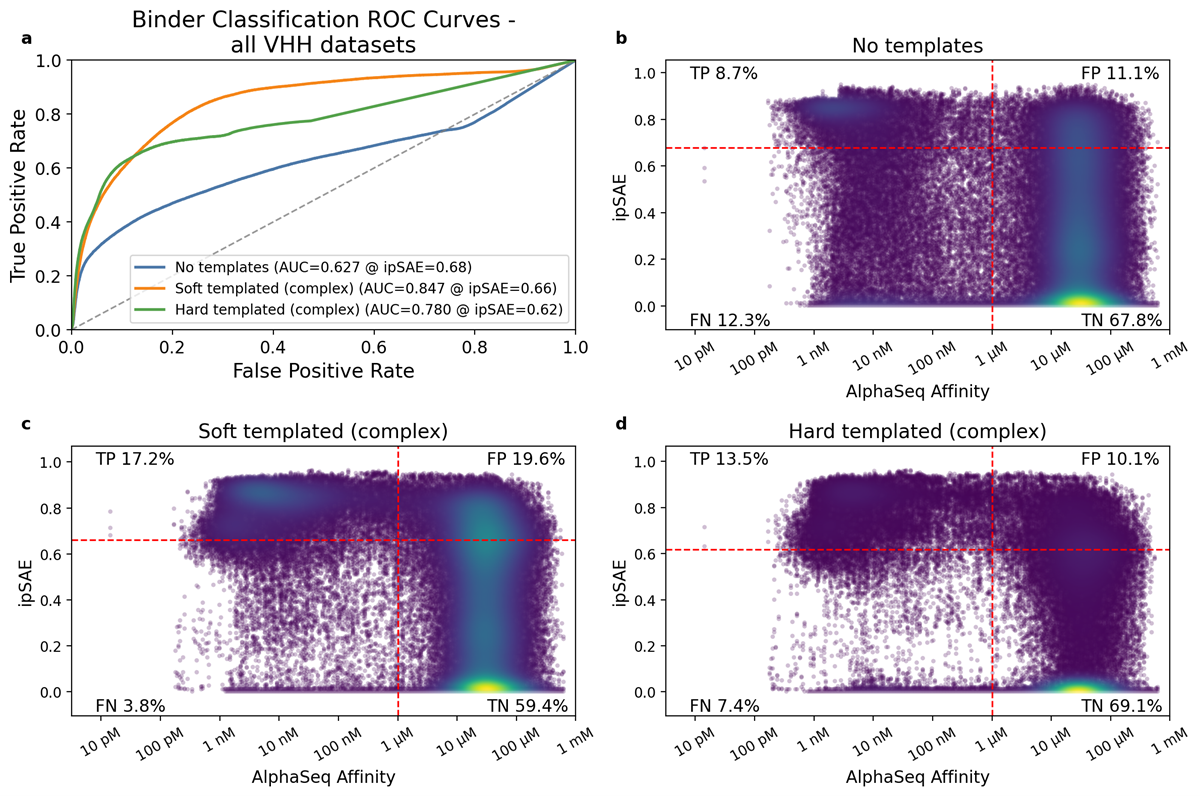
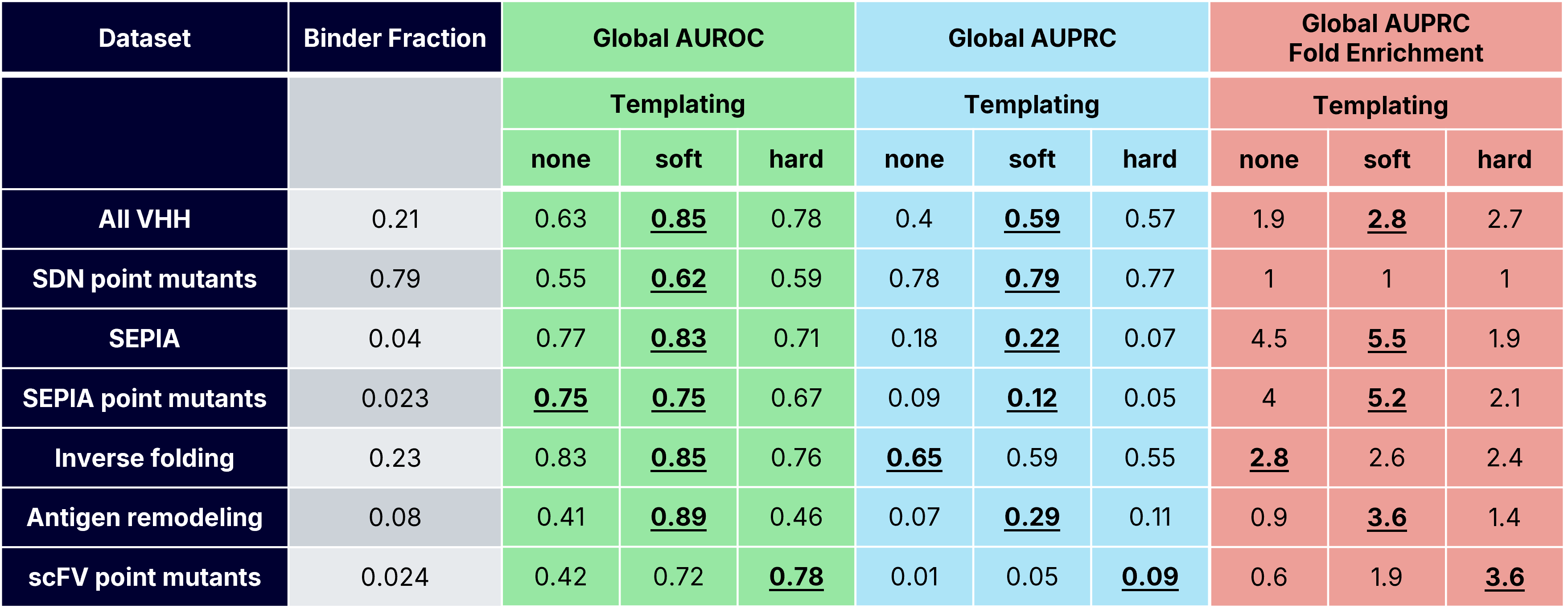
For each dataset, we evaluated how well ipSAE classified designs as binders (AlphaSeq affinity <1μM) or non-binders by computing the area under receiver-operator and precision-recall curves (AUROC and AUPRC). We also reported AUPRC fold enrichment, which is defined as AUPRC divided by the known positive binder fraction (Table 2). For each dataset, we defined an optimal ipSAE cutoff using Youden’s J-index (i.e. the threshold that best balances discrimination of binding/non-binding) [15].
ipSAE’s Performance
VHH Binder Classification
Across the four VHH-containing datasets, which consist of ~97,000 measured VHH-antigen interactions (Table 1), soft templating consistently outperformed both hard templating and no template strategies (Figure 2a, Tables 2 and 3). Soft templating achieved the highest true positive rate (17.2%) but also admitted the most false positives (19.6%). More broadly, ipSAE exhibited a persistently high false positive rate (>10%) across templating strategies. This finding reveals a consistent trade-off when using ipSAE as a binding classifier: a higher true positive rate also permits more false positives. In practice, ipSAE behaves primarily as a negative filter: it reliably removes many non-binders but struggles to cleanly separate strong binders from geometrically plausible failures.
VHH Point Mutation Classification
Mutation-level prediction remains challenging, as we and others have noted [14], with performance varying markedly across affinity predictions of point mutants. We compared two VHH datasets that introduce single mutations into VHH paratopes: SDN point mutants (VHH point mutants binding natural antigens) and SEPIA point mutants (VHH point mutants binding de novo minibinders). For SDN point mutants, ipSAE strikingly performed no better than random selection. In contrast, SEPIA point mutants showed strong binder enrichment (5.2X) for functional paratope mutants, comparable to the binder enrichment observed for the more sequence-diverse base SEPIA dataset (5.5X). One possible explanation is dataset context: SDN variants are derived from VHH–antigen structures present in the PDB that are present in Boltz-2 training data, whereas SEPIA interactions involve de novo designed minibinders that are absent from public structural databases. Together, these results suggest that ipSAE can detect mutation-driven functional changes in some settings, but with substantially lower reliability when compared to broader design tasks.
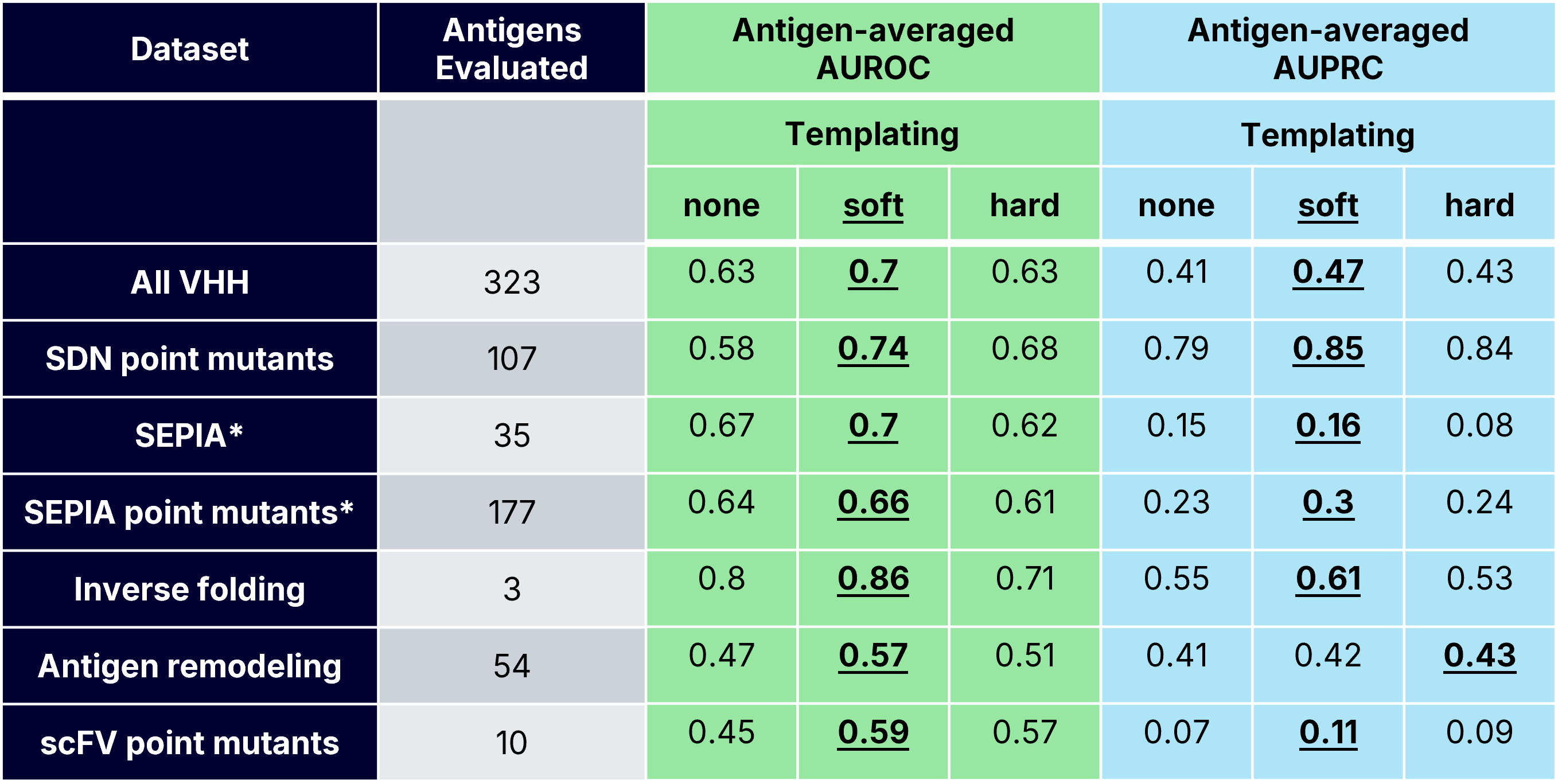
scFv Point Mutant Binding Classification
For the scFv mutant dataset, templating was critical for improving binder classification. Hard templating performed best globally, while soft templating performed best when averaging across antigens. However, soft and hard templating both showed variability, possibly due to the antigen monomer structure being used as the template for this dataset, as opposed to a full complex. These results reveal a practical lesson: when co-structures are unavailable, locking in antigen geometry (with a known or predicted monomer structure) can recover useful confidence signal. Without structural priors, ipSAE approaches noise.
Remodeled Antigen Binding Classification
Addressing challenging antigens is often critical in protein engineering, where truncated or stabilized variants are required to enable antigen purification, yeast display, or screening. This was the motivation behind our “Remodeled Antigens” dataset. It contained computationally remodeled antigens binding to VHHs, scFvs, and native binding partners with known co-crystal structures. Soft templating again performed markedly better than other templating strategies (Tables 2 and 3). Strikingly, soft-templated predictions yielded a 3.6x binder fold enrichment (Table 2). These results reveal that for substantially modified antigens, soft-templated complex predictions provide the most reliable discrimination between binders and non-binders and are more robust than either enforcing a strict structural prior or providing no structural prior at all.
Increased Confidence Requires Affinity Data at Scale
Across tens of thousands of PPI measurements spanning diverse systems of engineered binders and antigens, several patterns emerge.
First, structure prediction confidence scores are useful, but they do not perfectly differentiate between binders and non-binders and are ineffective at predicting relative binding affinities. Metrics like ipSAE enrich for binders, but many high-confidence binders fail when tested experimentally. In practice, confidence scores function best as negative filters: effective at removing poor designs, unreliable for ranking the best ones.
Second, structural context strongly influences performance. Providing templates during Boltz-2 prediction markedly improves binder classification, with soft templating consistently outperforming both hard constraints and no templates. Predictions made without structural priors produce confidence scores that poorly classify binders, while hard templating helps primarily in cases where geometry must be tightly enforced.
Lastly, mutation-heavy campaigns expose the limitations of confidence scores. Confidence scores often struggle to detect the subtle energetic changes that determine whether a single mutation improves or weakens binding, particularly for larger or structurally complex antigens. Performance varies by antigen, likely reflecting biases in training data, with familiar targets possibly yielding more reliable confidence scores than unfamiliar ones.
Large, quantitative datasets make these patterns visible. At scale, we observe overconfident non-binders, hallucinated interfaces, and antigen-specific blind spots. These results suggest that ipSAE and related metrics primarily capture bulk geometric plausibility rather than the energetic differences that determine binding affinity. The variability in confidence score performance for classifying binders is not so much a failure of structure prediction, but rather a mismatch between what confidence scores measure and how they are often used.
For protein designers, there are several practical takeaways:
Use confidence scores to filter out poor designs, not to rank binding strength.
Provide structural templates whenever possible. Even antigen monomer structures help.
Exercise caution when applying confidence metrics to mutation-heavy optimization campaigns.
Expect performance to vary by antigen. Better performance may be expected for antigens that are better represented in training data.
Looking forward, the path to more reliable confidence scores likely depends on the training data. Current confidence models were not trained on large, quantitative affinity measurements – they were trained on structural databases that capture only a relatively small, biased sampling of experimentally known co-complexes, with no information on how tightly they bind. Closing this gap will require explicitly training confidence models on datasets that span both highly diverse sequence and structure space, as well as local sequence perturbations. Large-scale affinity datasets of the kind described here represent exactly this opportunity: not just a benchmark for evaluating today’s tools, but a resource for building better ones.
Acknowledgements
Joseph Harman, Aditya Agarwal, Nick Altieri, Adrian Lange, David Noble, Kerry McGowan, and Natasha Murakowska performed the experimental design for these datasets. Davis Goodnight, Emily Engelhart, Mackenzie Goodwin, Shyam Gandhi, Kenny Herbst, Juliana Barrett, Charles Lin and Mimi McMurray contributed to the lab experiments. Joseph Harman, Natasha Murakowska, Nick Altieri, David Noble, Kerry McGowan, Adrian Lange, and Drew Duglan contributed to this work and text.
References
[1] Bennett NR, Coventry B, Goreshnik I, et al. Improving de novo protein binder design with deep learning. Nat Commun 2023;14:2625. https://doi.org/10.1038/s41467-023-38328-5.
[2] Wicky BIM, Milles LF, Courbet A, et al. Hallucinating symmetric protein assemblies. Science 2022;378:56–61. https://doi.org/10.1126/science.add1964.
[3] Bennett NR, Watson JL, Ragotte RJ, et al. Atomically accurate de novo design of antibodies with RFdiffusion 2024. https://doi.org/10.1101/2024.03.14.585103.
[4] Watson JL, Juergens D, Bennett NR, et al. De novo design of protein structure and function with RFdiffusion. Nature 2023;620:1089–100. https://doi.org/10.1038/s41586-023-06415-8.
[5] Pacesa M, Nickel L, Schellhaas C, et al. One-shot design of functional protein binders with BindCraft. Nature 2025;646:483–92. https://doi.org/10.1038/s41586-025-09429-6.
[6] Nabla Bio, Biswas S. De novo design of epitope-specific antibodies against soluble and multipass membrane proteins with high specificity, developability, and function 2025. https://doi.org/10.1101/2025.01.21.633066.
[7] Chai Discovery Team, Boitreaud J, Dent J, et al. Zero-shot antibody design in a 24-well plate 2025. https://doi.org/10.1101/2025.07.05.663018.
[8] Mille-Fragoso LS, Wang JN, Driscoll CL, et al. Efficient generation of epitope-targeted de novo antibodies with Germinal 2025. https://doi.org/10.1101/2025.09.19.677421.
[9] Swanson E, Nichols M, Ravichandran S, et al. mBER: Controllable de novo antibody design with million-scale experimental screening 2025. https://doi.org/10.1101/2025.09.26.678877.
[10] Stark H, Faltings F, Choi M, et al. BoltzGen: Toward Universal Binder Design 2025. https://doi.org/10.1101/2025.11.20.689494.
[11] Dunbrack RL. Rēs ipSAE loquuntur : What’s wrong with AlphaFold’s ipTM score and how to fix it 2025. https://doi.org/10.1101/2025.02.10.637595.
[12] Overath MD, Rygaard ASH, Jacobsen CP, et al. Predicting Experimental Success in De Novo Binder Design: A Meta-Analysis of 3,766 Experimentally Characterised Binders 2025. https://doi.org/10.1101/2025.08.14.670059.
[13] Cotet T-S, Krawczuk I, Stocco F, et al. Crowdsourced Protein Design: Lessons From the Adaptyv EGFR Binder Competition 2025. https://doi.org/10.1101/2025.04.17.648362.
[14] Pak MA, Markhieva KA, Novikova MS, et al. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLOS ONE 2023;18:e0282689. https://doi.org/10.1371/journal.pone.0282689.
[15] Ruopp MD, Perkins NJ, Whitcomb BW, et al. Youden Index and Optimal Cut‐Point Estimated from Observations Affected by a Lower Limit of Detection. Biom J 2008;50:419–30. https://doi.org/10.1002/bimj.200710415.