
Pairing Large-Scale Binding Affinity Measurements with Antibody-Antigen Structures
By David Noble

At a Glance: Datasets that combine structure, sequence, and affinity are highly valuable for training and evaluating models that predict antibody performance. But for VHHs, this data is limited: fewer than 100 of the ~1000 known non-redundant VHH-antigen complexes include measured binding affinities. To address this gap, we used AlphaSeq to measure binding affinities between hundreds of VHHs, VHH variants, and antigens from solved structures in SabDab-nano, yielding a total of ~2 million on- and off-target binding affinity measurements. The resulting dataset maps solved VHH-antigen structures to millions of binding affinity measurements, enabling us to interrogate strong on-target binding, local perturbations to known interactions, and the full scope of off-target interactions in a comprehensive dataset. In this post, we demonstrate the complexity and nuance in the landscape of VHH-target interactions and discuss how this rich dataset can be used for rigorous zero-shot antibody discovery and optimization workflows.
The Challenge of Antibody Engineering
Engineering clinically relevant antibodies is, at its core, an exercise in traversing high-dimensional fitness landscapes. Training models that can learn relevant features in this complex space will benefit from extensive, high-quality datasets, ideally with both structural and affinity labels. However, most antibody-antigen datasets illuminate only one or two aspects of the problem (sequence, or structure, or function), are often small, lack antibody or antigen diversity, or aggregate multiple assay outputs with inconsistent conditions [1–7].
Why Current Data Falls Short
Recent modeling advances have shown that multi-modal approaches can design functional antibodies. However, algorithmic innovation is outpacing the data being generated in antibody design; the bottleneck now is the scarcity of high-quality training data [2, 7, 8]. Antibodies exacerbate this challenge: unlike enzymes or natively evolving interacting proteins, they lack strong co-evolutionary signals in sequence databases, so traditional approaches are less informative for paratope–epitope recognition.
As of October 2025, structural coverage of VHH–antigen complexes is limited (<1,000 non-r-redundant entries, <100 with quantitative affinity) [1]. Sequence repositories such as OAS contain orders of magnitude more repertoires but are decoupled from antigen identity and lack functional ground truth [9]. Variant-level datasets such as deep mutational scans are uneven: biased toward a handful of parental antibodies and antigens, heterogeneous in assay readouts, and rarely providing matched negatives or cross-reactivity profiles. Synthetic data can extend but not replace real, diverse measurements.
Developing generalizable antibody ML models will require diverse, comprehensive data [2]. Robust design depends on these models not only learning features of strong binders, but also learning factors that impair binding – yet rich datasets that capture both positive and negative examples under consistent conditions remain scarce.
Our Dataset: Systematically Measuring VHH–Antigen Space
To address this gap, we used AlphaSeq to measure binding affinities for ~2 million antibody–antigen interactions across two assays:
Comprehensive coverage of VHH–antigen interactions from SAbDab-nano, including both on-target complexes and off-target mismatches (about 500K interactions).
Targeted mutational scans across CDRs for a subset of VHHs, introducing interface diversity with quantitative affinity labels (an additional 1.5M interactions).
AlphaSeq’s core strength is multiplexed, library-on-library, quantitative readout of pairwise protein–protein interactions under consistent, reproducible conditions [10]. This produced a dense matrix of VHHs × targets, where each interaction is a quantitative binding measurement. Figures 1 and 2 illustrate the global binding landscape and local mutational fitness landscapes measured in these assays respectively.
Measuring on- and off-target binding in SabDab-nano
Binding affinities for 261 antigens against 763 VHHs – for a total of 200,000 interactions, are shown in the heatmap below (Figure 1). This experiment reveals both expected on-target specificity between known interacting VHH:antigen pairs, as well as off-target interactions. Along the diagonal, we see strong, high-affinity signals where VHHs engage their intended antigen targets. A few off-diagonal signals also emerge: horizontal streaks reflect antigens bound by multiple VHHs, while vertical streaks indicate promiscuous VHH binding. Some off-target interactions are expected, including VHHs known to bind species homologs of their target. Others are novel or unexpected. These off-target effects underscore the intricate complexity of antibody-antigen recognition: even single-domain antibodies may exhibit cross-reactivity depending on structural similarity of the epitope or target of interest. The library-on-library nature of AlphaSeq enables us to quantify these interactions, providing extensive off-diagonal data for us to feed into antibody engineering models.
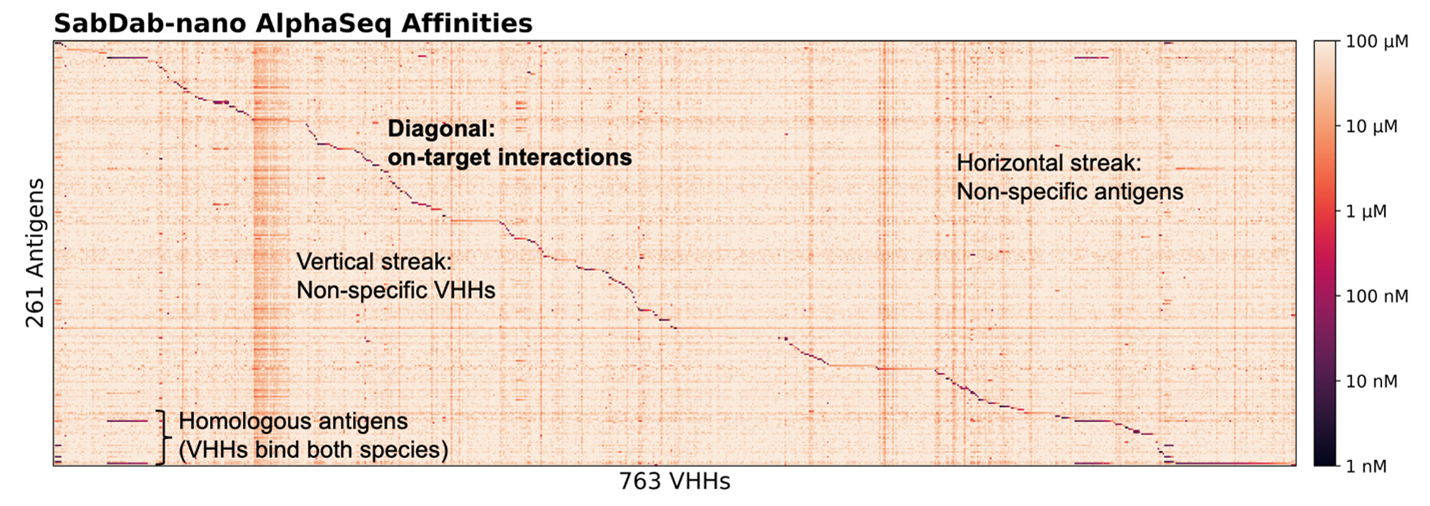
Figure 1 illustrates binding specificity and, in some cases, promiscuity. In a follow-up AlphaSeq experiment, we generate VHH point mutants against their intended targets to examine the sensitivity of these interactions to small perturbations. By introducing point mutations in each VHH, we observed substantial differences in binding relative to the parent VHH:antigen interaction. We probe this difference by measuring the delta-affinity (ΔKd), or change in binding affinity of a VHH variant compared to its parent VHH against the intended target. A negative ΔKd, indicates improved binding, while a positive ΔKd reflects weakening binding. The results are striking; while many variants bind weaker than their parents, a surprising subset achieve stronger binding than expected (best ΔKd <= -2.0, worst = 4.54, average = 0; AlphaSeq affinity scores are in log10 space). From a design perspective, this highlights many untapped opportunities to improve affinity.
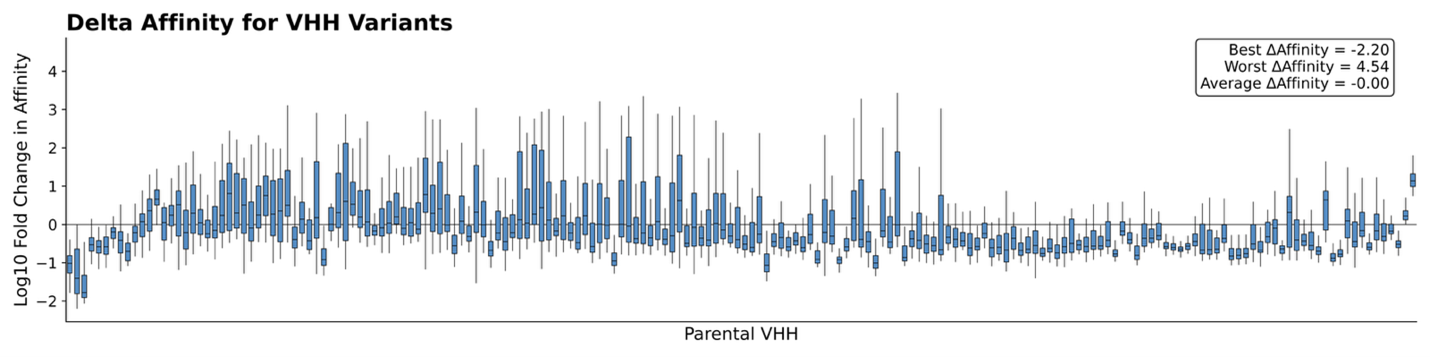
Taken together, these two experiments emphasize the dual nature of antibody engineering. On one hand, VHHs can exhibit broad cross-reactivity, binding to multiple related antigens. However, they can simultaneously be remarkably sensitive to subtle sequence changes – where a few mutations may either abolish or unexpectedly enhance binding. This paradox underscores the need for comprehensive characterization: the antibody fitness landscape is highly nuanced, and we can capture these complexities through systematically profiling the space with AlphaSeq.
What Makes Our Dataset Unique
This dataset is distinct in several ways:
Multimodal: Each paratope–epitope pair is annotated with sequence, parent structure (if on-target), and quantitative binding affinity.
Captures negative space: Off-target interactions are explicitly measured, revealing specificity as a distribution rather than a binary.
Reduced noise: Thousands of interactions measured in a robust assay with consistent internal standards eliminates much of the hidden heterogeneity present in public datasets.
Rigorous evaluation splits: With both structural and functional labels, we can define leakage-free splits by binder, by target, by complex, and by epitope class.
Shifting the Paradigm to “Complete” Multimodal Data
“Complete” multimodal data requires broader diversity across targets, epitopes, and scaffolds. Ground truth crystal structures provide a detailed view into the interaction between a paratope-epitope relationship, but only define “positive” strong binding interactions. To develop robust models, we will need these models to accurately pinpoint the difference between on-target versus off-target interactions, thus enable our models to learn what constitutes to a poor PPI interaction. We are expanding along these axes, including generating synthetic complexes validated in vitro. The goal is not just volume but coverage of biologically meaningful variation.
By providing models with comprehensive multimodal antibody-antigen data, we shift the learning problem from detecting weak correlations to learning causal constraints implied by the joint distribution of sequence, structure, and function. We believe this shift will yield models that are more sample-efficient, interpretable, and actionable.
Acknowledgements
Aditya Agarwal performed the bulk of the experimental design for these datasets. David Noble, Natasha Murakowska, Joseph Harman, Kerry McGowan, Nick Altieri, Adrian Lange, and Drew Duglan contributed to this work and text. Mackenzie Goodwin, Shyam Gandhi, Kenny Herbst, Juliana Barrett, Charles Lin and Mimi McMurray contributed to the lab experiments.
References
Schneider, Constantin, Matthew I J Raybould, and Charlotte M Deane. SAbDab in the Age of Biotherapeutics: Updates Including SAbDab-Nano, the Nanobody Structure Tracker. Nucleic Acids Research 50, no. D1 (2022): D1368–72. https://doi.org/10.1093/nar/gkab1050.
Matsunaga, R. & Tsumoto, K. Accelerating antibody discovery and optimization with high-throughput experimentation and machine learning. J. Biomed. Sci. 32, 46 (2025). https://doi.org/10.1186/s12929-025-01141-x
NaturalAntibody. AbDesign Database — Database of point mutants of antibodies with associated structures reveals poor generalization of binding predictions from machine learning models. (2025). Available at: https://www.biorxiv.org/content/10.1101/2025.06.09.658639v1
Jain, T. et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl Acad. Sci. USA 114, 944–949 (2017). https://doi.org/10.1073/pnas.1616408114
Zhao, X. et al. Benchmark for Antibody Binding Affinity Maturation and Design. arXiv 2506.04235 (2025). https://doi.org/10.48550/arXiv.2506.04235
Chungyoun, M., Ruffolo, J. A. & Gray, J. J. FLAb: Benchmarking deep learning methods for antibody fitness prediction. bioRxiv (2024). https://doi.org/10.1101/2024.01.13.575504
Hummer, Alissa M., Constantin Schneider, Lewis Chinery, and Charlotte M. Deane. Investigating the Volume and Diversity of Data Needed for Generalizable Antibody–Antigen ΔΔG Prediction. Nature Computational Science, Nature Publishing Group, July 8, 2025, 1–13. https://doi.org/10.1038/s43588-025-00823-8.
ProteinBase by Adaptyv: https://www.adaptyvbio.com/blog/proteinbase/
Tobias H. Olsen, Fergus Boyles, Charlotte M Deane. Observed Antibody Space: A Diverse Database of Cleaned, Annotated, and Translated Unpaired and Paired Antibody Sequences. Protein Science. October 29, 2021. https://pmc.ncbi.nlm.nih.gov/articles/PMC8740823/
Younger, David, Stephanie Berger, David Baker, and Eric Klavins. High-Throughput Characterization of Protein–Protein Interactions by Reprogramming Yeast Mating. Proceedings of the National Academy of Sciences 114, no. 46 (2017): 12166–71. https://doi.org/10.1073/pnas.1705867114